What is the study of Phylogenetics?
Phylogenetics is the study of the evolutionary relationship among biological entities [1]. A phylogenetic framework reveals the patterns of evolution of many morphological and chemical characters, including complex pathways[2].These relationships are discovered through phylogenetic inference methods with computer algorithms ranging in methods such as: distance matrix , neighbor joining, and Fitch Margoliash [3]. Phylogenetic trees can be constructed from “genetic distances” among species by aligning genomic sequences and measuring distance as number of mismatches between aligned positions.
Phylogenetics is the study of the evolutionary relationship among biological entities [1]. A phylogenetic framework reveals the patterns of evolution of many morphological and chemical characters, including complex pathways[2].These relationships are discovered through phylogenetic inference methods with computer algorithms ranging in methods such as: distance matrix , neighbor joining, and Fitch Margoliash [3]. Phylogenetic trees can be constructed from “genetic distances” among species by aligning genomic sequences and measuring distance as number of mismatches between aligned positions.
How to Build a Phylogenetic Tree
1) Create a FASTA formatted protein homology document
Creating a list of protein homologs from various related species showing conserved amino acid sequences with the human gene of interest allows for further experimental designs. Creating an edited FASTA formatted list also paves the way for creating different types of phylogenetic trees using programs such as MEGA, ClustalW2, and Phylogeny.fr.
Creating a list of protein homologs from various related species showing conserved amino acid sequences with the human gene of interest allows for further experimental designs. Creating an edited FASTA formatted list also paves the way for creating different types of phylogenetic trees using programs such as MEGA, ClustalW2, and Phylogeny.fr.

2) Create a CLUSTAL multiple sequence alignment
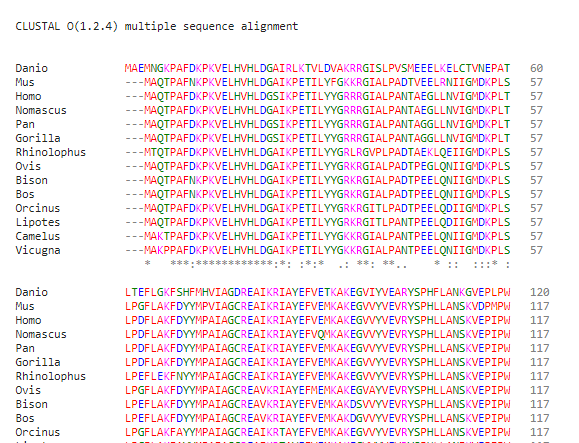
3) Create a Phylogenetic Tree with multiple methods
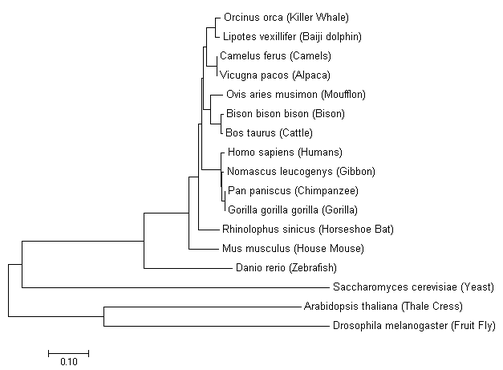
Figure [1] is a neighbor-joined phylogenetic tree without distance correlations created through MEGA software tools[5][6][7].
|
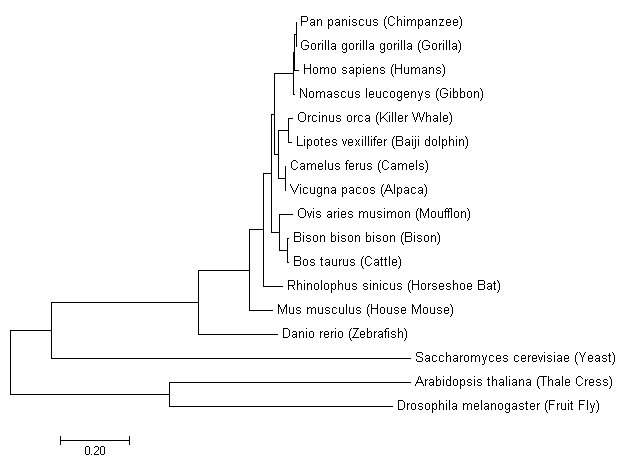
Figure [2] is a maximum likelihood phylogenetic tree without distance correlations created through MEGA software tools [5][6][7].
|
Tree Building Methods
|
Neighbor Tree Joining
Simlarity scores are calculated based off percent identity and with the scored determines which species are closer related for the creation of the tree. Branches are all different lengths for this method as opposed to other [2]. |
Maximum Likelihood
The main idea behind phylogeny inference with maximum likelihood is to determine the tree topology, branch lengths, and parameters of the evolutionary model that maximize the probability of observing the sequences at hand. The likelihood function is the conditional probability of the data (i.e. sequences) given [4] |
The branch of phylogenomics (no pun intended) is an important discipline of molecular biology, evolutionary genetics and many other fields for understanding how genes and genomes have evolved. It's a powerful tool to predict how these genes will continue to change in the future. Concluding on the very close lineage ADA shares with humans and the mice helps reinforce my previous selection of using it as a model organism to continue investigating the pathways involved with the mice ADA isoform.
Resources
[1] https://www.megasoftware.net/
[2]http://genetics564.weebly.com/homology--phylogeny.html
[3]https://www.ebi.ac.uk/Tools/services/web/toolresult.ebi?jobId=clustalo-I20180316-042006-0697-69626062-p1m&showColors=true&tool=clustalo
[4] http://www.ihes.fr/~carbone/MaximumLikelihood2.pdf
[5] Saitou N. and Nei M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4:406-425.
[6] Zuckerkandl E. and Pauling L. (1965). Evolutionary divergence and convergence in proteins. Edited in Evolving Genes and Proteins by V. Bryson and H.J. Vogel, pp. 97-166. Academic Press, New York.
[7] Kumar S., Stecher G., and Tamura K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets.Molecular Biology and Evolution 33:1870-1874.
[1] https://www.megasoftware.net/
[2]http://genetics564.weebly.com/homology--phylogeny.html
[3]https://www.ebi.ac.uk/Tools/services/web/toolresult.ebi?jobId=clustalo-I20180316-042006-0697-69626062-p1m&showColors=true&tool=clustalo
[4] http://www.ihes.fr/~carbone/MaximumLikelihood2.pdf
[5] Saitou N. and Nei M. (1987). The neighbor-joining method: A new method for reconstructing phylogenetic trees. Molecular Biology and Evolution 4:406-425.
[6] Zuckerkandl E. and Pauling L. (1965). Evolutionary divergence and convergence in proteins. Edited in Evolving Genes and Proteins by V. Bryson and H.J. Vogel, pp. 97-166. Academic Press, New York.
[7] Kumar S., Stecher G., and Tamura K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets.Molecular Biology and Evolution 33:1870-1874.
